BLAST Workflow
BLAST is a widely-used bioinformatics tool for comparing primary biological sequence information, such as the amino-acid sequences of different proteins or the nucleotides of DNA sequences with sequence databases, identifying sequences that resemble the query sequence above a certain threshold. The work performed by the COMPSs Blast workflow is computationally intensive and embarrassingly parallel.
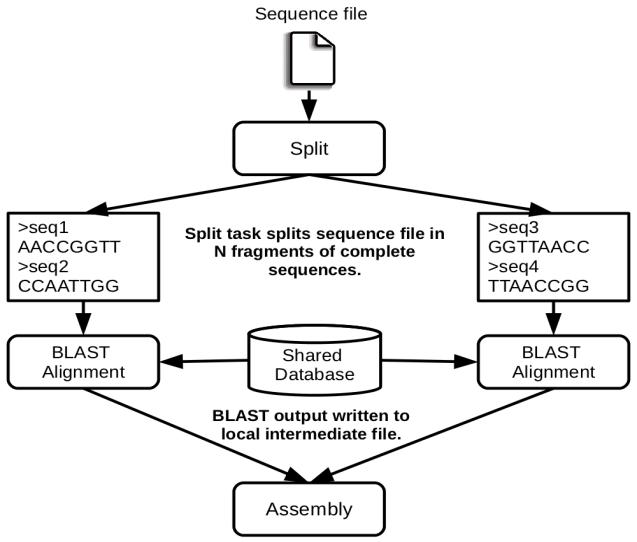
Figure 60 The COMPSs Blast workflow
The workflow describes the three blocks of the workflow implemented in the Split, Align and Assembly methods. The second one is the only method that is chosen to be executed remotely, so it is the unique method defined in the interface file. The Split method chops the query sequences file in N fragments, Align compares each sequence fragment against the database by means of the Blast binary, and Assembly combines all intermediate files into a single result file.
This application uses a database that will be on the shared disk space avoiding transferring the entire database (which can be large) between the virtual machines.
compss@bsc:~$ cp ~/workspace/blast/package/Blast.tar.gz /home/compss/
compss@bsc:~$ tar xzf Blast.tar.gz
The command line to execute the workflow:
compss@bsc:~$ runcompss blast.Blast <debug> \
<bin_location> \
<database_file> \
<sequences_file> \
<frag_number> \
<tmpdir> \
<output_file>
Where:
debug: The debug flag of the application (true or false).
bin_location: Path of the Blast binary.
database_file: Path of database file; the shared disk /sharedDisk/ is suggested to avoid big data transfers.
sequences_file: Path of sequences file.
frag_number: Number of fragments of the original sequence file, this number determines the number of parallel Align tasks.
tmpdir: Temporary directory (/home/compss/tmp/).
output_file: Path of the result file.
Example:
compss@bsc:~$ runcompss blast.Blast true \
/home/compss/tutorial_apps/java/blast/binary/blastall \
/sharedDisk/Blast/databases/swissprot/swissprot \
/sharedDisk/Blast/sequences/sargasso_test.fasta \
4 \
/tmp/ \
/home/compss/out.txt